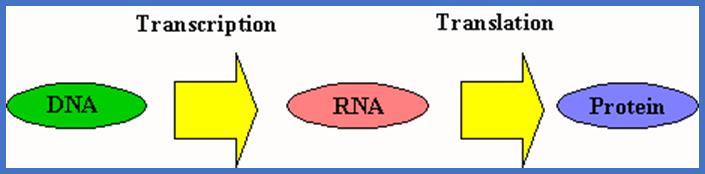
MOLECULAR BIOLOGY
The study of genes, its chemistry, structure, inheritance, expression and its regulation of the same at molecular level can be grossly called as Molecular genetics.
The material basis of inheritance was discovered by F. Miescher from pus cells collected from a hospital bandage cloth. The undigested material in the pus cells, as it is derived from the nucleus it was then called Nuclein. The nuclein was obtained from variety of sources as white crystalline powder, and chemically it showed acidic prosperities, so the Nuclein was called Nucleic acid.
Biologists and biochemists worked on this material extracted from variety of sources, and concluded that there are two types of Nucleic acids, one from the plants ex. yeast and another from animals, ex calf thymus.
But Feulgen demonstrated by using what is now called Feulgan’s reagent, that there two types of nucleic acids, irrespective of the sources, may be from plants, or from animals or from microbes, they are Feulgan’s positive called Deoxy Ribose Nucleic acid and another Feulgan’s negative called Ribose Nucleic Acid.
F. Griffith, using streptococcus pneumoniae strains like virulent encapsidated bacteria and rough non-virulent strains, showed that heat killed virulent strains can easily transform living but non-virulent strains when both were injected to the same mice. Thereby he established the presence a genetic transforming principle in virulent strains.
By that time the presence of chromosomes as vehicle heredity was firmly established and Mendelian Genetics has its impacts on heredity and the importance of genes to specific characters and the interaction of them in expression of characters was firmly established.
Yet the material basis of inheritance was still an elusive substance.
Meanwhile Chargaff worked on quantitative analysis of nucleotide bases from a variety of sources, showed that the quantity of purines were found to be equal to that of total amount of pyrimidines and the total amount of Adenine was found to be equal to total amount of Thymine and total amount of Cytosine was found to be equal to total amount of Guanine. This analysis was stemmed from the DNA sources of microbes and eukaryotes.
Avery, Macleod and McCarty using streptococcus strains clearly established that DNA isolated from virulent alone was capable of transforming non virulent strains of bacteria, when bacterial strains are incubated with extracted DNA.
This was further confirmed beyond doubt by Hershey and Martha Chase, where they used radio active isotopes, for labeling proteins (14C leucine) and nucleic acids (32p Uridine) of T2 bacteriophages one with the labeled proteins and another with labeled DNA and infecting bacteria separately and tracing which goes in and which molecules replicate and produce viral particles.
Though chemical basis of inheritance was established beyond doubt, the existence certain viruses such as Tobacco Mosaic Viruses, were able infect, multiply, mutate and perform certain functions in generating few protein has made people to realize, that the material basis of inheritance is just not the monopoly of DNA; but RNA can also act as genetic material.
It is now firmly established that any organic molecule which can replicate, mutate, recombine and perform certain functions is called as the Genetic Material or the material basis of inheritance.
Chemistry of Nucleic Acids:
Chemically both Deoxy ribose and Ribose nucleic acids are made up of a) five carbon pentose sugar with 1’ beta hydroxyl group, b) a phosphoric acid (H2PO4^+2), and c) nitrogen bases such as purines (Adenine, Guanine) and pyrimidines (cytosine, thymine and uracil).
DNA invariably consists of Deoxy ribose sugar i.e., oxygen moiety at 2’ carbon is absent, while ribose contains OH at the same position.
With respect to bases, DNA contains Adenine, Guanine, Cytosine and Thymine and uracil is not at all found, where as RNA contains Adenine, Guanine, Cytosine and Uracil. Very rarely, Thymine is present in RNA as ribo-thymidine.
Covalent linking of sugar with a base by a glycosidic bond between C1 of the ribose and the 1’N of pyrimidines or 9’N of purines of the bases produce Nucleosides.
Addition of Phosphate group to the 5’carbon of the sugar makes them Nucleotides. In most of the cases the tri phospho nucleotides act as precursors of DNA and RNA.
Nucleotides of DNA and RNA are named on the basis of their bases present, such as Adenylic Acid, Guanylic acid, Cytidilic acid, Thymidylic acid and Uridylic acid. Nucleotides for DNA are generally called dNTPs and for RNA they are called as rNTPs.
Structurally whether it is DNA or RNA, they are made up of poly nucleotide strands, where nucleotides are covalently bonded between 3’ OH group of sugar of one nucleotide with that of phosphate group found at 5’th carbon of the second nucleotide. The chemical linkages generate what is called Phospho diester bond.
This dinucleotide has a polarity, i.e., a free 5’ triphosphate group in the first nucleotide and free 3’OH group in the second nucleotide.
Addition of any more nucleotides always take place at the free 3’OH group of the second nucleotide, again by another phosphodiester bond formation. Like this any number of nucleotides can added to form a long chain of ploy-nucleotides.
The addition and the formation of polypeptide chain is always pre-existing templates and it is performed by specific enzymes.
So far, no enzyme has been found which can add nucleotide by 5’ end.
The kind of polynucleotide chains form depends upon the kind of nucleotides they incorporate into the chains; it can be DNA or it can RNA.
Structurally and functionally each of them differs and have their own characteristic features.
DEOXY RIBOSE NUCLEIC ACID (DNA)
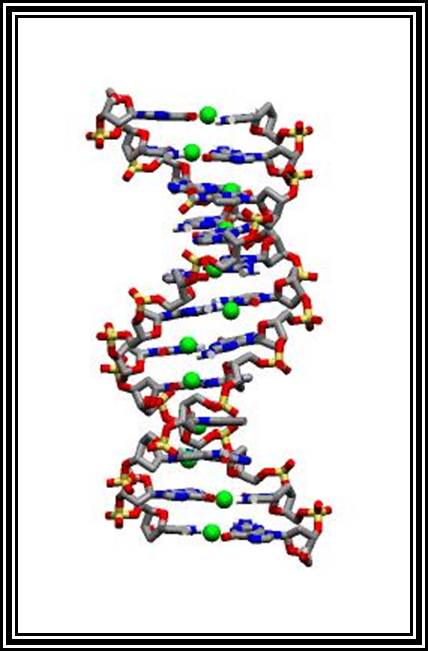
In most of the organisms DNA is the genetic material. It is a long molecule with a molecular weight ranging from 105 to 1012 daltons. The size and the amount of DNA in a cell is always constant and species specific. The content of DNA varies from species to species. It is very stable and not subjected rapid turnover. It has all the information in encoded form required for the organism's structure and development. The genomic DNA is not restricted to some viruses, prokaryotes and eukaryotes; such genomic DNA is also found in cell organelles such as mitochondria and plastids.
DNA molecules exist in various forms such as single stranded, double stranded; triple stranded or four stranded or quadruplex form. They can exist either in linear or circular form, but all these forms are species specific.
Structure of single stranded DNA:
A single stranded DNA when suspended in aqueous media, the strand takes random route, yet while in water it exhibits random motions with right-handed turns and twists . It has polarity with 5’phosphate group at one end 3’ OH group at the other end. The sugar phosphate back bone shows smooth curvature. The nucleotides are bound to sugars at right angles to the long axis of the backbone. The bases whether purines or pyrimidines are made up of heterocyclic rings, flat, rigid because of double bonds and they are hydrophobic in nature. The bases with respect to sugar phosphate axis show slight tortional twist. This stems for the fact the bases with respect to sugars are in anti configuration for the sugars show C2’ endo and C3’ exo folding or sugar puckering. The bases are one above the other and exhibit hydrophobic interactions; hence the propeller twist to the single stranded DNA. Wherever complementary sequences exist the ssDNA base pairs and exhibits secondary structure, with hair-pin structures, or stem loop structures or if the length of double strand region is longer it can exhibit helically coiled structure.
Structure of double stranded DNA:
B-DNA:
· It consists of two polynucleotide strands of opposite polarity.
· They are helically coiled to each other because of their base pairing between Adenine and Thymine; and Guanine and Cytosine.
· This kind of base pairing follows what is now called Chargaff’s rule.
· Specificity of base pairing between the said bases is actually due to the prevalence of specific ionic forms of the bases in the cellular milieu. The ionic state or tautameric state of Adenine and Cytosine in amino form and not in iminoform.
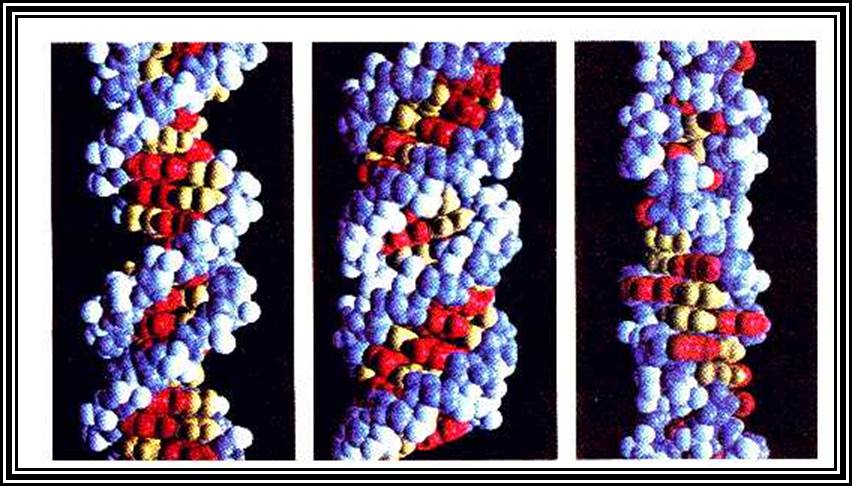
Molecular Model
DNA right-handed helix
On the contrary the guanine and thymine exist in ketonic form than enol form. It is these features that drive the DNA to undergo base pairing only between Adenine and Thymine; and Guanine and Cytosine. Under normal conditions any other base pair combination is just not possible. But under certain structural situation like triple stranded form of DNA or quadruplex form one set base pairing will be Watson-Crick base pairing and the other is non-Watson-Crick or Hoogsteen base pairing. The pairing is due to hydrogen bonds; two hydrogen bonds between Adenine and Thymidine and three hydrogen bonding between Guanine and Cytidine, because that is the one and the only possibility.
· In the double stranded structure, bases are at right angle to long axis of sugar-phosphate back bone.
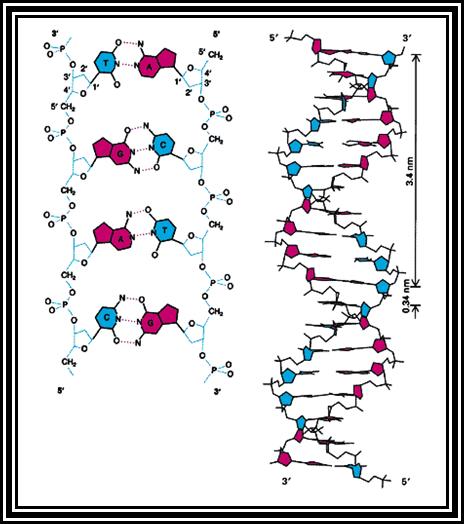
Diagram showing structural features of DNA.
· The bases with respect to sugars are in anti-configuration for the sugar puckering is C2’ is endo and C3’ is exo.
· The rigid, planar and hydrophobic bases are at an angle to the axis.
· When they base pair to each other they are not exactly parallel and planar to each other but they are in little angularly oriented. This provides the angular twist or what is called propeller twist.
· This force is further rein forced by the hydrophobic interaction between successive base pairs.
· All these features contribute to the DNA to take right-handed helicity with specific dimensions.
· Though hydrogen bonding is force to recon with, hydrogen bonding perse provide specificity of complementarity of base pairing and stability to DNA helix stems from planar base pair stack’s hydrophobic interactions.
· The DNA molecule exhibits right-handed helix called B-helix, it has a diameter of 20A^0.
· For every 10.6 bp, it takes one complete turn.
· The sugar-phosphate backbone shows smooth curvature.
· The major groove shows 22A^o width and the minor groove has 12 A^o size.
Z-DNA:
· Some regions of DNA show left-handed helix with a diameter of 18^A.
· Its sugar-phosphate back bone takes a zig-zag path. This is due to the base’s orientation with respect to their sugar stereo-structures.
· This type DNA structure is possible only when the sequences in the DNA are CGCGCG and its repeats. If the Cs in the CG sequences are methylated, the bases are forced into syn orientation to the sugar, for the sugars are in C2’exo and C3’in endo puckering,
Structurally different forms of DNAs- Helical, and Zig-Zag.
· This configuration drives the axis to take zig-zag orientation and also it takes left-handed helicity.
· The diameter of the helix is 18A^o and it takes 12 bp for one complete turn.
· Such DNA is called Z-DNA.
· The Z-DNA structural form is found only small segments and specific regions such as promoter loci. Both B-DNA and Z-DNA can structurally and immunologically be distinguished.
· The B-DNA is the major structural form and Z-DNA exists as small segments with in the B DNA form. However, the Z-DNA is G-C rich.
· The nucleotide sequence of any DNA molecule is unique and it acts as the language of life.
Other forms of DNA:
Depending upon the sequences, DNA can assume variety structural forms. The DNA does not or need not have the same B-DNA structure throughout its length. Based on the sequences and the repeat of specific sequences, it can assume varietal structural forms such as, Bent-DNA, Flexible-DNA, Curved -DNA, Supercoiled DNA, Triple stranded-DNA, and Quadruplex-DNA and others.
Triple stranded DNA:
Depending upon the sequences and the length of repeat sequences, the DNA can take triple stranded form, where two strands are base paired according to Watson-Crick base pairing and the third strand normally gets into the major groove and base pairs with Hoogsteen base pairing. Such triple stranded forms can be due to intra strands or inter strands base pairing. Such abnormalities have been detected in certain diseased patients.
Quadruplex DNA:
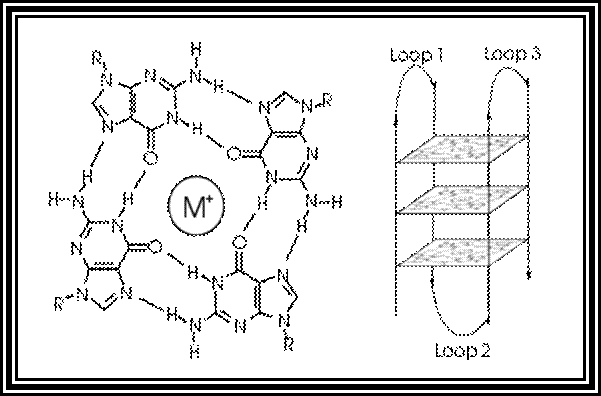
Certain sequences rich in Gs can assume quadruplex form. Very good example is from Telomeric DNA. Telomeric DNA has certain sequences repeated several hundred times at the terminal region of the DNA. For example, in Tetrahymena Telomeric DNA, its sequence of TTGGGG. In such situations every second G base pair with second G of the next segment. Thus, it can form what is called quartet structural form. Such structures are stabilized by the binding of Telomeric specific proteins.
In molecular biology, G-quadruplex secondary structures (G4) are formed in nucleic acids by sequences that are rich in guanine.[2] They are helical in shape and contain guanine tetrads that can form from one,[3] two[4] or four strands.[5] The unimolecular forms often occur naturally near the ends of the chromosomes, better known as the telomeric regions, and in transcriptional regulatory regions of multiple genes, both in microbes[6][7] and across vertebrates [8][7] including oncogenes in humans.[9] Four guanine bases can associate through Hoogsteen hydrogen bonding to form a square planar structure called a guanine tetrad (G-tetrad or G-quartet), and two or more guanine tetrads (from G-tracts, continuous runs of guanine) can stack on top of each other to form a G-quadruplex.
The placement and bonding to form G-quadruplexes is not random and serve very unusual functional purposes. The quadruplex structure is further stabilized by the presence of a cation, especially potassium, which sits in a central channel between each pair of tetrads.[3] They can be formed of DNA, RNA, LNA, and PNA, and may be intramolecular, bimolecular, or tetramolecular.[10] Depending on the direction of the strands or parts of a strand that form the tetrads, structures may be described as parallel or antiparallel. G-quadruplex structures can be computationally predicted from DNA or RNA sequence motifs,[11][12] but their actual structures can be quite varied within and between the motifs, which can number over 100,000 per genome. Their activities in basic genetic processes are an active area of research in telomere, gene regulation, and functional genomics research.[13][14]
WIKIPEDIA
Super coiled DNA:
Whether ssDNA or ds DNA, circular or linear most of the DNA exists in supercoiled form. When the DNA, because in certain situations, where the DNA is over twisted or under winded, in such situations, they are subjected to certain strains and stresses. Thermodynamically feasible base pair per helix is 10.6. If the DNA is over wound in right handed, the number of base-pairs per turn will decrease, which is thermodynamically not acceptable, so in order to accommodate such extra turning the DNA under goes coiling on its own length, similar to that of an a rubber band turn on it own ring. Similarly, under wounding situations the numbers of base pairs increase from the normal 10.5 bp. In order to over come, the DNA undergoes super coiling. The right-handed twist on its own is called positive super coiling; this is due to over winding of the DNA. If the DNA is under wound the super coiling on the right-handed direction, which is called negative super coiling.
Supercoiled DNA can be easily detected by running the DNA in an Agarose gel. If a supercoiled DNA and relaxed DNA of the same size is run on a gel, the supercoiled DNA runs faster than the relaxed DNA for the supercoiled DNA is compact.
These super coils are removed or added all the time. There are specific enzymes called Topoisomerases such as Topoisomerase I and TopoisomeraseII or called Gyrase are the enzymes found in Prokaryotes. Such Topoisomerase are also found in Eukaryotic systems.
Replication of DNA:
Duplication of DNA in its entirety to generate two exact copies at molecular level is called Replication. It is one of the fundamental, or call it, cardinal principle or properties of life. The replication has to be absolutely perfect to the core and it cannot afford to make even a single mistake. Even if such mistakes are made, they should be repaired immediately. This is exactly what happens during replication.
Meselson and Stahal demonstrated that the process of replication of ds DNA is principally a semi conservative process, where, the each of the two daughter molecules will posses or retain one of the two parental strands, which acts as the template for the synthesis of complementary strand. Even single stranded DNAs also exhibit similar feature, where the parental strand, whether it is (+) or (-) strand, acts as the template for the new strand. And the new strand acts as the template for another strand, then one of the strands is packed into viral capsid proteins.
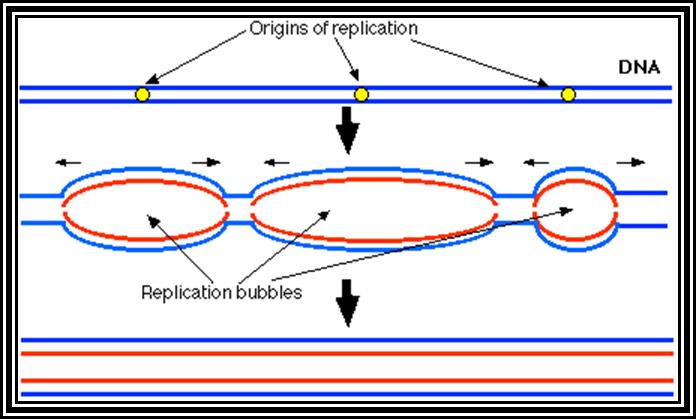
Basically replication, whatever may be the type of DNA, replication starts or initiates at a predetermined site called Origin (ORI) of replication. And the replication ends or terminates at predetermined site called TER site. A DNA consisting of an origin and a TER site is called a Replicon. For example, E. coli genomic DNA has the size of 4.6X10^6 bp is considered as replicon. Even little larger genomes consist of one replicon and it takes about 8 to 10 minutes to replicate at the rate of 50,000 to 60,000 bases per minute. On the contrary eukaryotic chromosomes contain genomes of the size ranging from 10^7 to 10^8 bp. And the time period required for replication is hardly 6-8 hrs. If the rate of replication in Eukaryotes is 2500 base per minute, the time required for the replication of the entire chromosome requires about 40,0000 minutes or 6666.6hrs or 277 days if it continuously moves from ne end to the other end, Hence, replication mechanism in EK has employed multiple origins in a single chromosomal DNA; if all the origins fire simultaneously the replication of the entire chromosome can be completed in a limited time.
Whether it is PK or EK DNA, the process of replication involves three stages, one Initiation, second Elongation and third Termination. Each of the events requires specific factors and specific enzymes, the factors and the enzyme required for pK and EK are different. Even viral DNA employs different factors and enzymes. The following description of structure of origin and factors and the mechanism is prokaryotic DNA.
Factors required for Initiation:
|
Gene |
Protein |
Mol.wt (KD) |
Function |
|
dna-A |
Dna-A |
50 |
Binds to Dna-A box |
|
dna-B |
Dna-B |
50 x 6 |
Helicase |
|
ssB |
SsB |
19 x 4 |
Bind to s-p-s backbone |
|
top-I & II |
Top-I &II |
100 and 97 |
Relaxing super coils |
|
Dna-G |
Dna-G |
60 |
Primer laying |
Factors required for Elongation:
|
Gene |
Protein |
Mol.wt (KD) |
Function |
|
Pol-A |
DNA-pol I |
103 |
Primer removal and gap filling |
|
pol-E |
DNA-pol alpha subunit |
132 |
5’>3’ polymerase |
|
dna-Q |
Epsilon |
27 |
3’>5’ exonucleases |
|
Hol-E |
Theta |
10 |
Facilitates complex assembly |
|
Dna-X |
Tau |
71 |
Facilitates dimerization of core complex |
|
Dna-X, hol-A, hol-B,hol-C, hol-D, |
Gamma complex(delta, delta’,chi,psi |
52x2,35,33,15,12 |
He3lps in loading the beta clamps |
|
Dna-N |
Beta subunit |
37x2 |
Holds DNA and Pol. |
|
Ligase |
DNA-ligase |
75 |
Ligates the nicks |
Factors required for Termination:
|
Gene |
Protein |
Mol.wt (KDs) |
function |
|
tus |
|
23 |
prevents renitiation |
|
|
|
|
|
|
|
|
|
|
Origin:
Origin is the region at which replication is initiated. The name of the origin is based on the DNA in which is found, ex. Ori-C is found in E. coli DNA, ColE-1 is found in Col-E 1 plasmid, Ori-SV40 in SV-40 DNA (EK), Ori-lambda and Ori T7 in Lambda and T7 phage DNA.
In E. coli, the there is only one genome of a size of 4.6X10^6bp. Structurally it has one origin site at 84.5 mpu and one TER site exactly opposite to the origin.
Structurally the origin region is about 250 bp long and consists of block of consensus sequences like four 9- mers and three 13-mers; deletion of any of the nucleotide in the sequence hinders initiation.
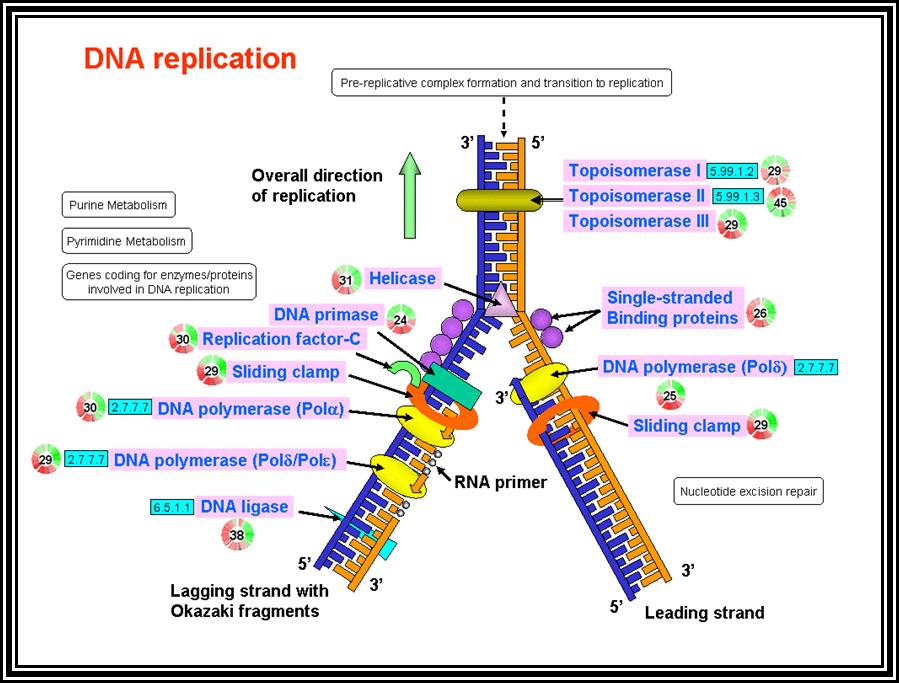
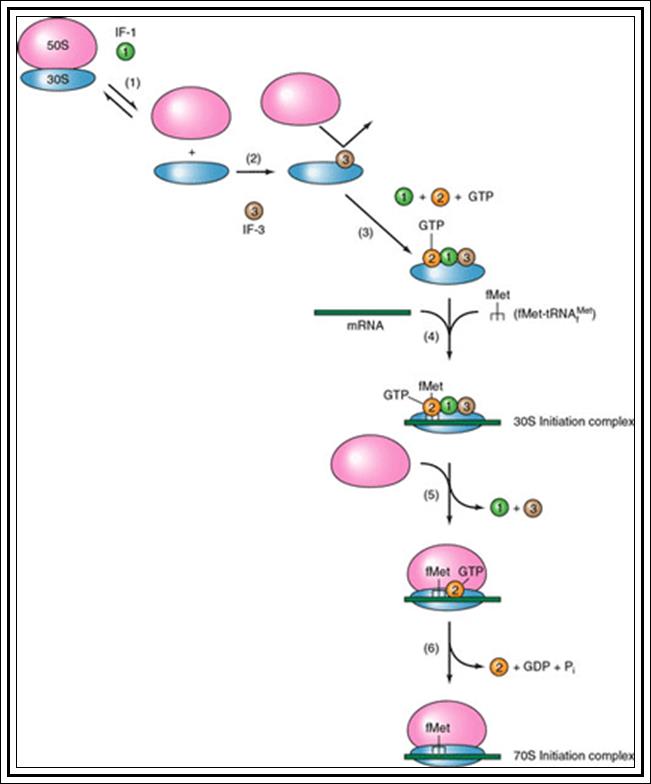
Initiation:
Just before initiation, due to full methylation (both strands) in the origin region (GATC), where ‘A’ is methylated, the circular DNA frees from the mesosomal membrane from the attachment point.
The gene DNA-A product Dna-A which is synthesized at the time of replication initiation, binds to each block of 9-mer sequences. Binding is co-operative and the proteins build up in the region. As a consequence, DNA wraps around these protein clusters. Wrapping of the DNA around the DNA-with proteins results is torsion that induces the DNA to melt and open in 13-mer region as single stranded structures which looks like a bubble called Replication eye.
The ssDNA strands are prevented from reannealing for single strand binding (SSB) bind to phosphate-sugar-phosphate backbone; it also provides stability and rigidity to the strand.
Almost at the same time Dna-B facilitated by Dna-C assembles on to one of the strands at fork joint as an hexamer ring around the ssDNA; it is an ATP dependent process. The Dna-B complex is a motor protein, it is ATP dependent Helicase. Using ATP, it can drive forward along the double strands open the DNA ahead of the fork.
The fork movement creates over winding of the DNA ahead of it, so in order to relax topoisomerases act on super coiled DNA and relax the super coils.
At the same time another protein called Dna-G joins the Dna-B hexamer. Dna-G, it is a primase, for it produces short 11 ntds long primers on lagging strand. The primer is a short piece of RNA which provides a kind of anchor with 3’OH, a site from which a nucleotide chain can be extended.
The complex Dna-B, Dna-G and other associated proteins can be considered as primosome.
Elongation:
As the replication bubble is loaded with primosome complex, DNA polymerase III complex along with accessory factors like gamma complex and beta clamps assemble at the fork joint.
The gamma complex consists of several factors, which facilitate the loading of beta clamps on to each of the strands at the base of the enzyme complex.
The beta subunits are clamp shaped proteins assemble on to single strands as clamps and also associate with polymerase complex. Thus, they prevent the polymerase to dissociate from the template. Thus, the processivity increases.
The DNA polymerase (holozyme) consists of core components such as Alpha subunit (actually it is the polymerase), Epsilon (3’->5’ exonuclease, provides proof reading activity), Theta (facilitate core complex assembly) and Tau (helps in core complex dimrization). This complex mainly responsible for the replication of DNA.
In the replication bubble there are two replication forks. Among the two strands at each fork, the one that is loaded with helicase and primase is considered as lagging strand and the opposite strand is called Leading strand. This is because the lagging strand replication is slightly delayed than the other strand.
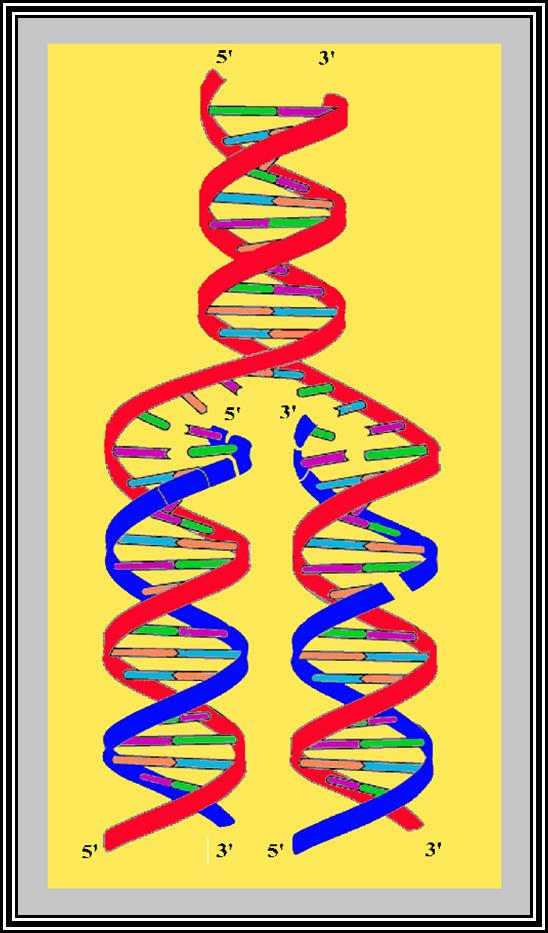
Figure shows replication initiation with new double stranded DNA forming.
This mode is possible when the replication is bi-directional, otherwise replication is unidirectional.
The assembly of DNA pol-III complex at each fork is in the form of dimers .
At each fork the lagging strand is in 5’->3’ direction and the DNA helicase moves only in 5’>3” direction on this strand.
Lagging Leading
<-3’----------------------------‘I----------------------------5’
5’---à
<-------5’
5’__________________ I’_________________>3’
Leading lagging
Primers are laid for both leading strand and lagging strands at each specific sites. Primers are laid on strands by primases, but the primer laid on the leading strand is believed to be from RNA pol. However, the primers laid on the lagging strand, are at an interval of 1000 to 1500 nucleotides, by the primase.
The DNA polymerase dimer complex associates one at each fork. The paradox is that the polymerase synthesizes the new strand only in 5’--.3’ direction and never in the opposite direction. The new complementary chain on leading is 5’--->3’.
As both polymerase complexes move in the same direction, the synthesis of new polynucleotide chain on lagging strand becomes a paradox for the orientation of the lagging strand itself in 5’-->3’ direction.
This enigma has over come by the explanation that during the synthesis of new chain on lagging strand the DNA is wrapped around in such a way;
the new strands are now oriented in opposite orientation and DNA-pol as it moves forward it uses the short primers laid on lagging strand extends the chain till the Pol meets another primer, at which place the Pol release from the new strand and moves on to the next primer and extends the chain.
The synthesis of new strand on leading is continuous and the synthesis on lagging strand is discontinuous, thus the short fragments synthesized are called as Okazaki fragments, named after the discoverer.
The presence of beta clamps associated with DNA-Pol explains why the procesessivity of the polymerase is so high. Procesessivity means the length of the new strand produced by the polymerase without dissociating from the template and fast. High processivity means longer strands are produced, though the synthesis of new strands on lagging strand is dispersive.
While the new strands are synthesized the helicase continuously forges ahead of the fork and makes the single strand available. The continuous movement of the motor protein in fork region is facilitated by the Topoisomerases, continuously relax the tightly coiled DNA ahead of the fork.
The RNA primers are removed by DNA polymerase, for it has both 5’-->3’ and 3’-->5’ exonucleases activity. As the polymerase also has 5’>3’ polymerase activity, it extends the 3’ end of the DNA and fills the gap.
The gap that is left is at the adjacent nucleotides, with 3’OH and 5’Phosphate groups.
The DNA ligase of the E. coli is an NAD dependent ligase. The enzyme binds to DNA and scans the DNA and where it comes in contact with such nicks, it performs covalent phosphodiester bonds, and thus the ligation takes place. Removal of the primers and sealing the nicks are continuously performed while DNA replication is in progress.
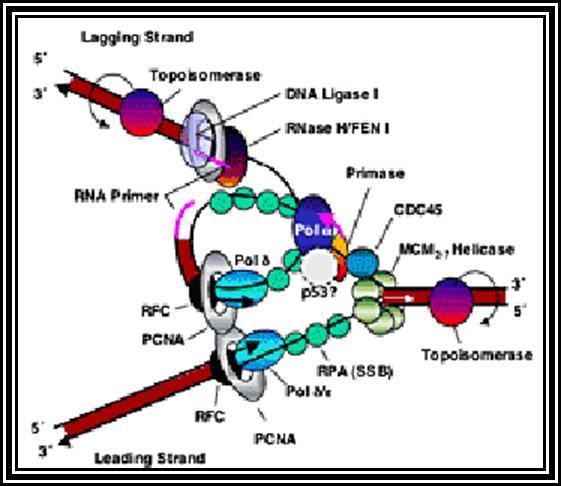
The DNA polymerase enzyme has the binding site for the template and another site for the primers end. It has catalytic site next to the 3’ end of the primer. The enzyme monitors the correctness of the complementary nucleotide, it is only the base, is geometrically complementary and fits into the site, the enzyme catalyzes the covalent bond formation, otherwise it is not. Even by chance the nucleotide assembled is not proper, the enzyme move back and remove the nucleotide by its inherent 3’-->5’ exonucleases activity.
The rate of synthesis is 50000 to sixty thousand per minute. This is the highest polymerizing activity for any other known polymerases.
Termination:
The replication which starts at a site moves in bidirectional with the same rate. As the replication forks approach each other at their end points, there is every possibility and probability the fork can move into the newly synthesized Ds DNA, thus a new round of replication possible. This is prevented for the TER regions contain three sets of TER sequences oriented in opposite directions. These sequences are recognized by TER associated proteins. Thus, when Helicase move into such TER sites the TUS substances bound prevent the movement and new round of replication is prevented. Though this true for E. coli DNA, such TER sequences are required for the completion of replication.
RIBOSE NUCLEIC ACID (RNA)
Ribonucleic acids (RNA) are another class of nucleic acids. In recent years, the role of RNAs in cellular metabolism, regulation of genes and gene products, growth and development of an organism, is gaining increased attention of molecular biologists all over the world.
With the exception genetic RNAs found in some viruses, all other RNAs play a secondary role to DNA in executing the functions directed by DNA. The amount of RNA in a cell varies depending upon the functional status of the cell. Resting cells have less amount of RNA, on the contrary dividing and active cells contain greater amount of RNA, which suggests the functional status of the cell. Most of the RNA is found in cytoplasm and the rest is found in the nucleolus and karyolymph. They are not stable and they are always subjected to rapid turnover. The half life of prokaryotic mRNA is hardly 2-5 minutes, while the mRNAs of eukaryotes is 10 to 15 minutes. But ribosomal and tRNAs are more stable. Their size varies from 67 ntds to 15000 ntds.
Chemical Composition: Similar to DNA, RNA is also made up of sugar called ribose. The phosphate group is same. Among the four N2 bases Adenine, Guanine and Cytosine are same, but Uracil is found instead of Thymidine.
The nucleotides of RNA are rAMP, rGMP, rCMP and rUMP. The Polynucleotide chain formation is similar to that of DNA. It also has a polarity i.e., 5' to 3' ends.
Structure: Structurally RNA molecules are made up of a single polyribonucleotide chain with 5’ and 3’ polarity. But they exhibit secondary and tertiary conformations. Few RNAs show double stranded features.
Types of RNAs: Depending upon the structure and function they are classified into Genetic or Genomic RNAs, Ribosomal RNAs (rRNAs), Transfer RNAs (tRNAs), Messenger RNAs (mRNAs), and small molecular wt RNAs (sn/sc RNAs), sno RNAs (C/D RNAs and H/ACA RNAs), TmRNAs, Si and Mi RNAs, Xist RNAs, Primer RNAs, Trans-acting RNAs and few more.
Genetic RNAs: They act as the genetic material. They exhibit replication, mutation, recombination and also involved in protein synthesis, eg. Tobacco mosaic viral (TMV) RNAs (+ ve sense), Picorna viral RNAs (+ve sense), Rhode viral RNAs (-ve) sense, Influenza viral RNAs (-ve sense), Roe viral RNAs (ds RNAs), Rous sarcoma viral (RSV) RNAs (+ RNAs), Human immunodeficiency viral (AIDs or HIV) RNA (+ sense), Human T-cell lymphocyte leukemia viral RNAs (HTLV, + sense) and many others including Viroidal RNAs and Virusoidal RNAs. Many viroidal RNAs consists of only one genome per virus, but some contain two similar genomes (HIV), some may contain 8 different genomic RNAs enclosed in the same viral capsid. On the other hand, Reo viruses contain ten different ds RNAs.
A distinguishing feature of Genetic RNAs is that they can replicate, recombine, mutate and perform functions in coding for proteins. No other types of RNAs have the said properties. Most of these RNAs are found as the genomic materials of viruses and viroids. All viruses are encapsidated in distinct coat proteins; some do contain host cell membrane as envelope over the capsid proteins.
Genetically many of the RNAs exhibit positive sense and others exhibit negative sense. Positive sense means the nucleotide sequence of them is similar to that of mRNA and they can be translated to generate proteins. On the contrary negative sense RNAs have sequences complementary positive strands. They cannot be translated, but they can be translated only when positive RNAs are produced on the negative strands. The genomic RNAs in general code for one protein as a polyprotein or they can code for more than one protein either by splicing or by generating different mRNAs. Some RNAs such as bacteriophage RNAs have structural features similar to that of prokaryotic, but majority of the genetic RNAs of plant and animals exhibit eukaryotic features. Most of the positive sense genomic RNAs have caps at their 5’ end (it is a 22 amino acid protein called VpG in the case of Picorna viral RNA, 7’methylated cap in the case of TMV and most of the positive sense RNAs have poly(A) tail.
Replication:
In general, positive sense RNA as the genomic material gets into the cytoplasm; it is subjected translation by the host machinery (retroviruses are exceptions to this rule). On the other hand, negative genomic RNA viruses carry pre-made replicase enzymes with them. When the viral genome is injected into the cell, viral enzyme is used for the replication of the negative strand. Some use protein mediated primers for their replication and some use tRNA or tRNA like structural conformation of the viral genome as primers.
In general, the positive stranded RNA is copied to negative strand, and then the same enzyme copies the negative sense RNA into positive stranded RNA, which is then packaged into capsid. In the same manner the negative sense RNA is copied into positive sense RNAs, then it is copied into negative sense RNA, then it is packaged into viral capsid. But retroviral RNA has positive sense, but it is not translated on delivery into cytoplasm, but it is copied into double stranded cDNA, this is in turn is transported into the nucleus, where the cDNA gets integrated into host chromosomal DNA, thus it can remain there without affecting the host. When such genome is activated, the viral DNA produces full length transcripts and also produces the required proteins, resulting in viral particles.
Ribosomal RNAs:
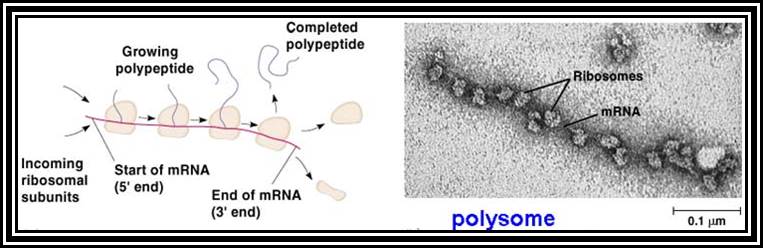
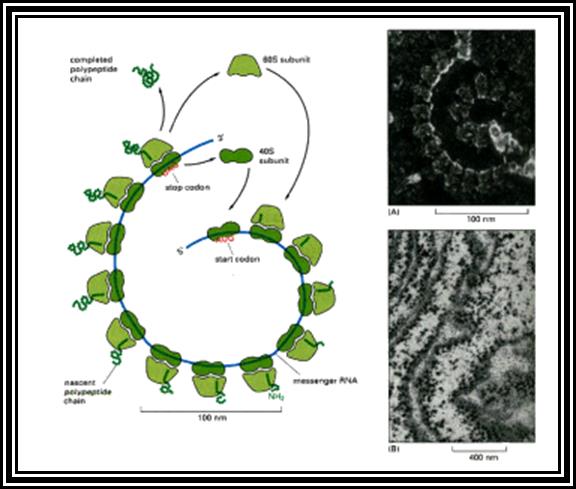
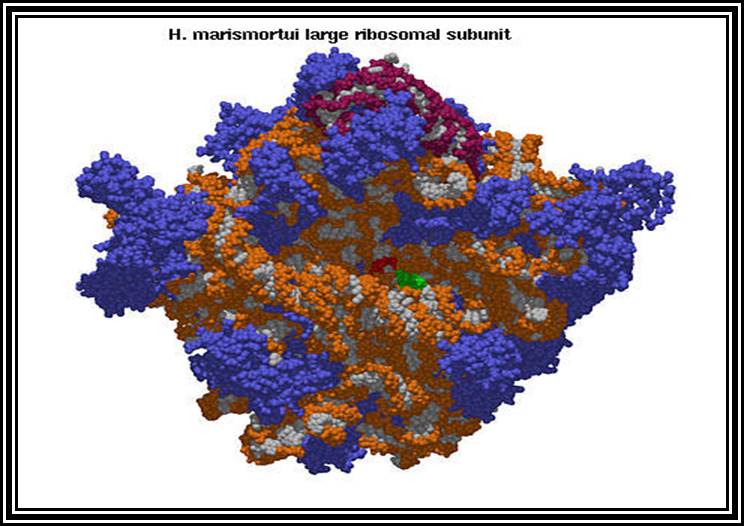
Ribosomal RNAs constitute the most abundant class of RNAs, where more than 90% of the total cellular RNA is ribosomal RNA. They are the constituents of ribosome. They are called structural RNAs and they are made up of different classes and sizes. In association with riboproteins, they organize into ribosomes. They have an important role in protein synthesis.
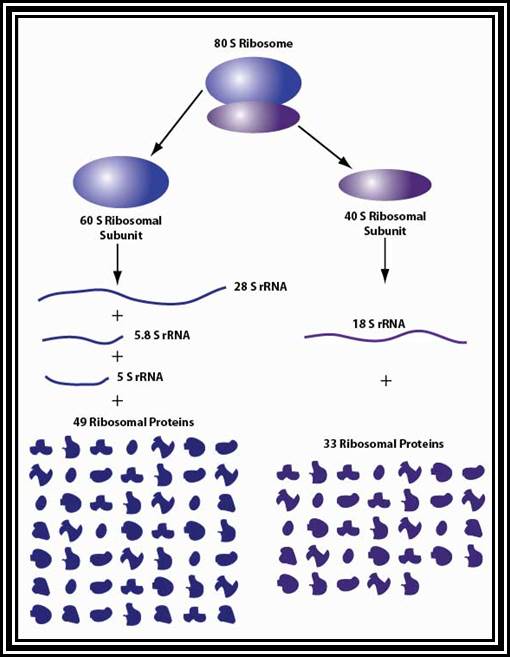
Ribosomes are the most important structural components of the cell in protein synthesis. Based on the size and molecular weight, two distinct class of ribosomes can be identified; one of Prokaryotes and the other as Eukaryote, however the eukaryote also contain prokaryotic like ribosomes in their cell organelles. Ribosomes of both PK and EK contain two subunits each, based on their sedimentation coefficient values they are named small and large subunits of specific ‘S’ value. Such as 70S = 50s+30 subunits, and 80S=60s+40 subunits.
5s r RNA- structural features
Molecular analysis of the moderately repetitive DNA shows the number of ribosomal RNA genes and riboprotein genes in biological systems.
70s ribosomes (Prokaryotic):
|
Subunits |
RNA size |
Riboproteins |
Methylation |
No. of genes |
|
50s subunits |
2900 ntds(23s RNA) 120 ntds(5sRNA) |
31; L1 to L31 |
20 at 2’OH of ribose sugars |
Seven genes for both, all RNAs together |
|
30S subunits |
1540-42 ntds(16s RNA) |
21: s1 to s21 |
10 at 2’Oh of ribose, 2 at Adenine and 2 at Guanines |
|
80 S Ribosomes (Eukaryotic):
|
Subunits |
Size of RNAs |
Riboproteins |
Methylation |
Gene numbers |
|
40s |
1843-1900 ntds (18s RNA) |
34: s1 to s34 |
43-44 at 2’OH of ribose |
200 to2000 for 40s/60s together |
|
60s |
4718-4800 ntds (28s RNA); 160ntds(5.8sRNA); 120ntds(5sRNA |
45-50: L1 to L50 |
74 at 2’OH of ribose sugars |
Number of genes for 5S RNA- 200 to 2400 |
Ribosomal structural features.
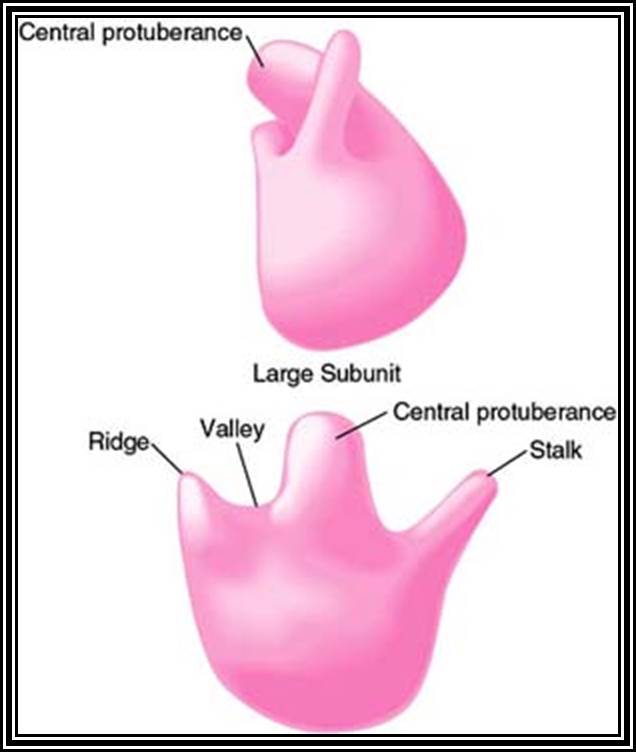
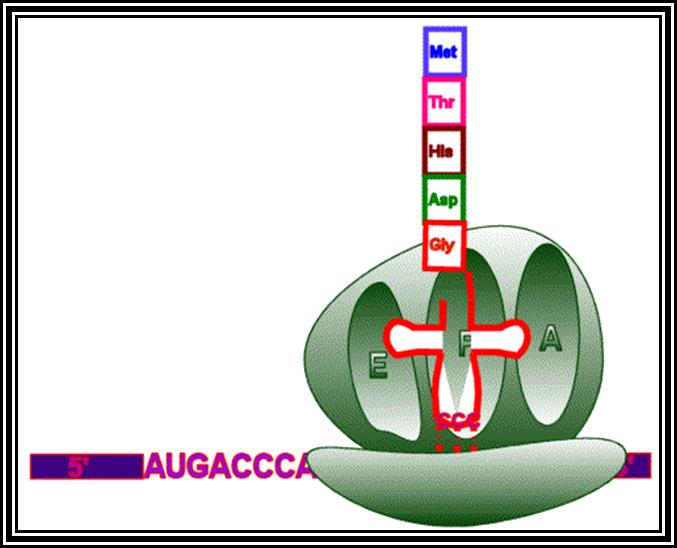
Ribosomes perse, whether they are 80s or 70s are submicroscopic particles of 100 x 220 A^o size. Small subunit shows head and a body, where the head is split into inner and outer flat structures. The space between them is called cleft. It is on the platform the tRNAs bind and through the cleft the mRNA threads through. It also contains sites for the binding aminoacyl tRNAs, one is called P-site (peptidyl-tRNA site) and the other is A-site (aminoacyl-tRNA site), next to the A-site there is another site called A’-site, where arriving tRNAs are screened for accuracy. In both pK and EK systems it is the small subunit that reacts first with the 5’ end of the mRNA. In prokaryotes the 16 RNA at its 3’ end has a sequence complementary to the sequence found in leader sequence of mRNAs, it is through base paring between them the mRNA binds to ribosomes. In EK the 5’end is bound by cap binding protein associated protein complexes. The rRNAs and proteins are associated in sequence specific manner in a 3-D model of the ribosome structure. The structure is not rigid, but vey flexible and dynamic in nature.
The large subunit is more or less appears as a palm with a cup-shaped at the base, and a central protrubence, an elongated projection and another blunt projection similar to the thumb at the sides. It is into this concave shaped structure, the small subunit tucks into during protein synthesis. The large subunit also contains P-site and A-site and also it has E-site (tRNA exit site) next to P-site. More to it, it has narrow groove for the protein elongation towards the base. It through the groove the newly synthesized protein threads through. The size of the groove is 20-25A^o and length it can accommodate about 40 a.a long protein chain. Many of the proteins bound to RNA are strategically positioned in such a way they have their own functions to perform
Most of the rRNAs are synthesized as large precursor RNAs, then they are sliced, processed and specifically methylated, and then they undergo folding into 3-D structure in association with riboproteins which associate rRNAs in sequential steps, to generate 3-D structure of the ribosome. Each of the ribosomal substructures has specific functional domains.
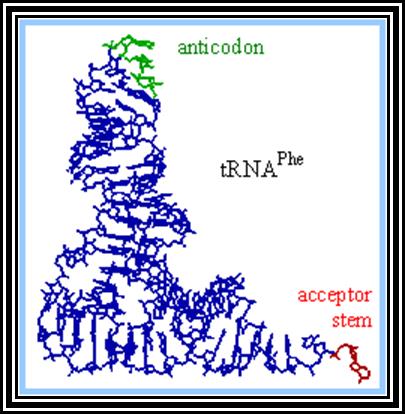
Transfer RNAs:
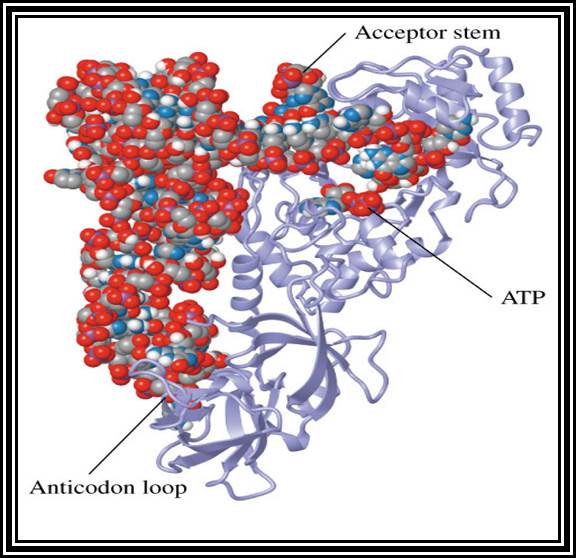
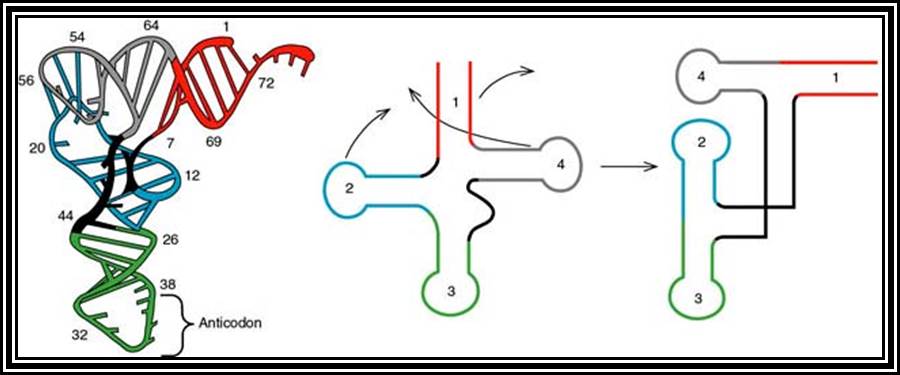
These are also called soluble RNAs. They are very small molecules and made up of single polynucleotide chains of 75 to 98 ntds long. They are found in cytoplasm. They exhibit 3-D structure. Sequence based secondary structure was first proposed by Holley, for which ha was awarded Nobel Prize. The model was based on it shape which is like a trifoliate leaf of a clover plant, hence it is called clover leaf model. Accordingly, the tRNAs consist of various domains from the 3’ end such as, 1) Aminoacyl-acceptor end, 2) TUCG loop U= pseudouridine), 3) Anticodon loop and 4) DHU (Dihydroxy Uridine) loop; each have base paired stem such as acceptor stem, TUCG stem, Anticodon stem and DHU stem respectively. In between DHU stem and anticodon stem there is a small extra loop consisting of 5 to 20 ntds. Amino acid acceptor end has binding site to aa-t.RNA synthase. TUCG loop has a function to bind to ribosomal surface. Anticodon loop binds to specific a.a t.RNA synthase and has a function in decoding. The DHU loop has a function in recognizing the aat.RNA synthase and gives specificity to amino acid and tRNA. Each of these regions has specific functions. Most of the tRNAs contain modified bases, such as pseudouridine, 3’ methyl or 5’methyl cytosine
3-D structure and secondary structure of tRNAs
dihydroxy uracil, isopentenyl adenine, inosine, wyosine. Such modifications are sequence specific and modifications take place only after the synthesis of tRNA precursor. The acceptor end has 3 unpaired ntds 3’ACC. The 5’ end of the tRNAs is invariably G paired with Cytosine. The anticodon loop consists of specific combination of triplet nucleotides which act as anticodons. The anticodons are codon specific. On either sides of the anticodon are bracketed by modified bases such as pseudouridine on the 5’ side and Isopentenyladenine or any purines at 3’ side. The anticodons are structurally organized in such a way they are geometrically and stearically disposed for accurate base pairing with codons found in mRNA, (which are disposed similarly when they are bound to ribosome and present their codon for base pairing).
Secondary structure is sequence model, but the tRNAs fold into tertiary structure into a compact and crystallizable structure. Such mode of folding greatly favored by the presence of specific modified bases. Modification of bases and specificity varies from tRNA to tRNA, but it is species specific. The compact folding is reinforced by tertiary base pairing, which is similar to Hoogsteen bonding or what is generally termed as non-Watson-Crick base pairing. When it is folded it exhibits an inverted L-shaped where the height and the tail length is 60A^o and the diagonal length between the tip of the tail and anticodon end is 70A^o. Though there are 61 codons, there are only 22 or 30 types of tRNAs.
In addition to the above number, one more tRNA has been added that is Selenocysteine tRNA for selenocysteine is the 21st amino acid in the genetic code directory. Still, they cognately recognize all the 61 codons by employing what is called third base wobble mechanism. In wobble mechanism, the first base of the anticodon wobbles i.e. the nucleotide found at the 5’ end, base pairs with the third base of the codon, it means G can pair with C or with U, and A can pair with U and Inosine can pair with U, A or C. Thiouracil can pair with Adenine or Guanine. During anticodon and codon reactions mRNA is read in 5’ to 3’ direction, so the codons are also in 5’->3’ direction. But the tRNAs base pair with codons in opposite orientation i.e., 3-’>5’ orientation. So, the first anticodon base in tRNAs is in 5’ position and the third base to mRNA, which is found as the last base in 3’ position. Interestingly biosystems don’t produce any tRNAs for any of the three nonsense or chain terminator codons. But some tRNAs have the ability to read through terminator codon and produce a protein.
3’-C.C.C-5’---tRNA: (anticodon)
5’---G.G.G-3’---mRNA: (codon)
Anti-codon first base: =: Codon third base
3 xxG 5’ = 5’ xxC 3’ normal
3’ xxG 5’ = 5’ xxU 3’ wobble
3’ xxA 5’ = 5’ xxU 3’ normal
3’ xxG 5’ = 5’ xxU( thiouracil) (wobble)
3’ xxA 5’ = 5’ xxU (wobble)
3’ xx I 5’ = 5’ xxC 3’( I= inosine) Wobble)
3’ xx I 5’ = 5’ xxU 3’ (wobble)
3’ xx I 5 ’= 5’ xxA 3’ (wobble)
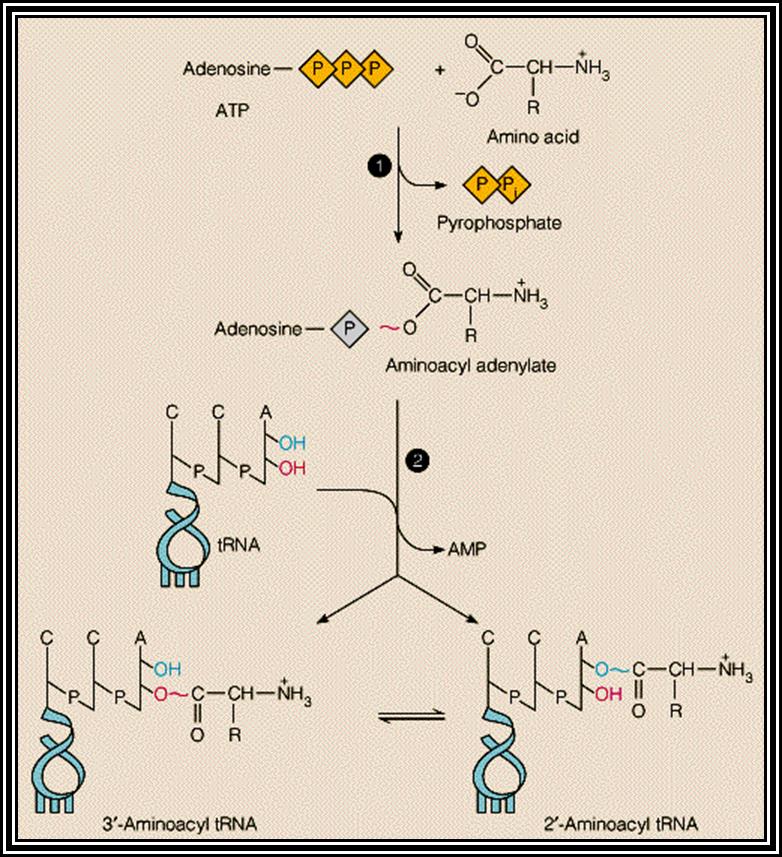
All amino acids, (there are 20000 or more in E. coli and more than a million in EK), bind to 3’A of ACC as a consensus sequence of tRNA. It is to the A nucleotide in all tRNA amino acids are covalently added. How, what amino acid to be added to which tRNA poses a jig-saw puzzle. However, a specific amino acid is loaded to a specific tRNAs. There are many tRNAs which have isoacceptors, which means two or more different tRNAs carry the same amino acid, similar to different codon sequences code for the same amino acid called degeneracy.
Specific Aminoacyl tRNA synthetase an enzyme adds amino acid to the terminal A nucleotide of the tRNA. This enzyme is amino acid and tRNA specific and their number depends on the number of tRNAs and amino acids.
The tRNAs carry amino acids by their amino-acceptor end. They are highly specific for amino acid residues, which is determined by their structure and the enzyme aminoacyl tRNA synthetase. There are twenty or more different kinds of tRNAs, which are structurally specific at least in their nucleotide sequence, but the 3’ terminal end is same in all tRNAs so also the number of enzymes.
Aminoacyl tRNA synthetase enzymes are the enzymes responsible for adding specific amino acid to specific tRNA. This recognition of specific tRNA, and specific amino acid and adding the amino acid residues to the same 3’AAC end is often called Second Genetic coding mechanism. All biosystems have encoded with specific amino acyl tRNA synthetases for each and every species of tRNAs. Such specificity stems from the fact the each of the enzyme has specific structural conformation with specific site for a specify amino acid and a specific tRNA. However, it has a common site for the AMP which is common for all enzymes and it is required for enzyme activation.
First, the enzyme activated by the binding of ATP to its ATP binding site, which results in the hydrolysis of ATP to AMP and PPi, where AMP remains bound to the enzyme. The activated enzyme now accepts the correct amino acid to form aminoacyl AMP. At the same time, it can also accept a specific tRNA. Reports clearly indicate that the sequences in acceptor stem, DHU loop and anticodon loop are considered as the recognizing sequences and it is to which the enzyme makes specific contact (binding). The binding of tRNA is in such a way the 3’ACC of the acceptor end is a place at a very close proximity to aminoacyl end. This leads to the formation of a covalent bond between the 2’ or 3’ OH group of the A with carbonyl group of the amino acid; high energy carbonyl group is covalently transferred to the A 2’ or 3’ OH group; still this carbonyl bond is an energy rich bond. This process is called tRNA charging. The binding of a. a change the conformation of the tRNA into conformationally active state.
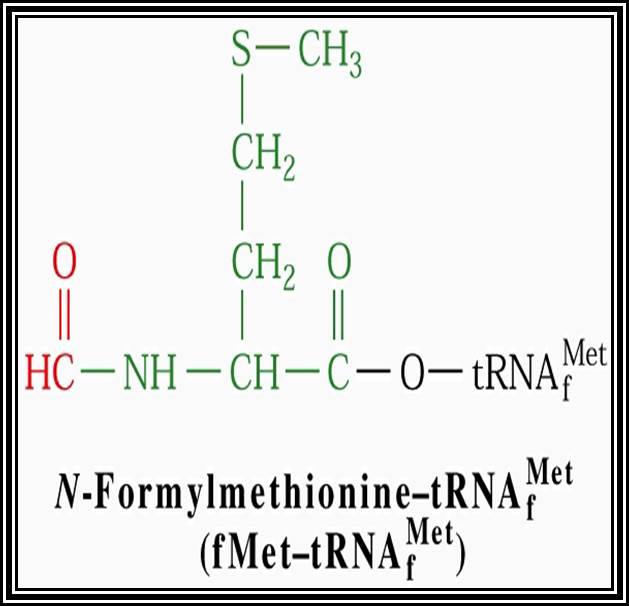
In general, most of the tRNAs are aminoacyl tRNAs, which are charged with specific amino acids. But there are specific tRNAs called initiator tRNAs, which are absolutely required for initiating protein synthesis. They are called Intiator tRNAs. In PK they are called as formyl-methionine tRNA and in EK they are methyl-tRNAs. They have specific structural features for specific a. a-tRNAs. These a.a tRNAs are placed on ribosomes bound to mRNAs complementary to their codon sequences. This is a remarkable specifity process.
Messenger RNAs:
They are single stranded polynucleotide chains and carry message in the form of codons for amino acids and they determine the sequence of amino acids of a polypeptide chain, which in turn determine the structure, and function of the protein. Structurally PK and EK mRNAs show certain distinct features.
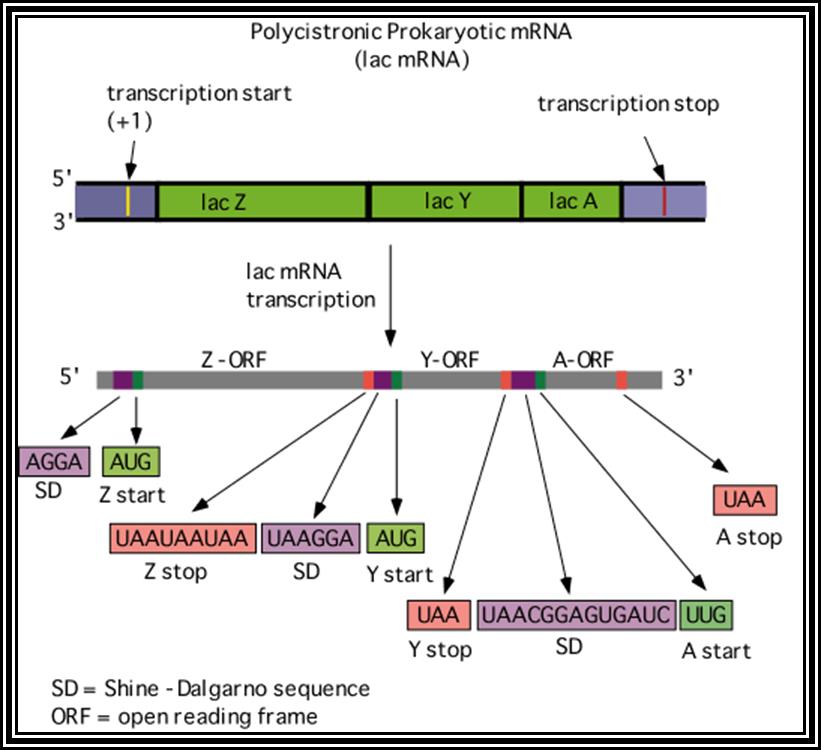
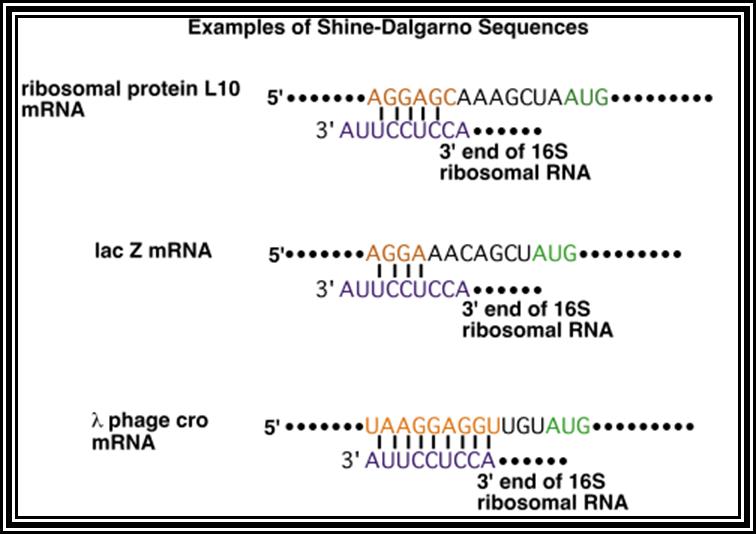
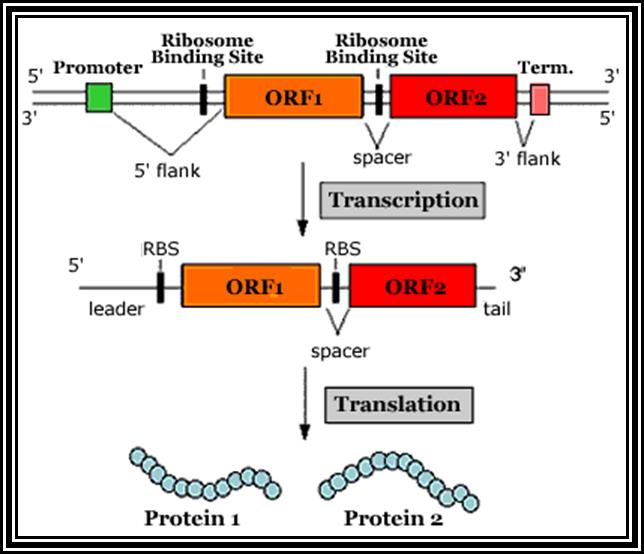
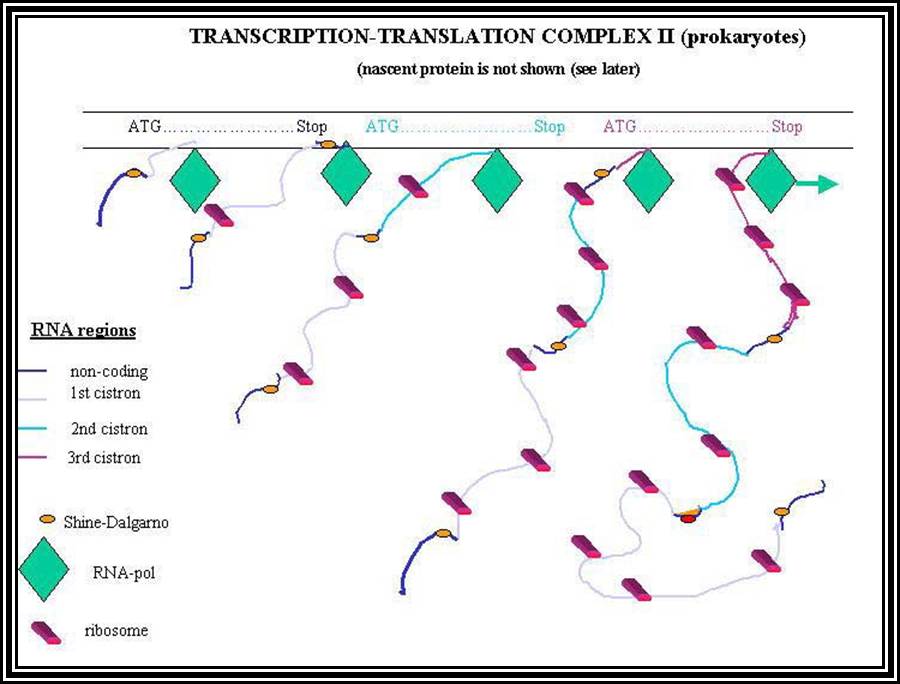
Prokaryotic mRNAs: As they are synthesized, they are brought on to ribosomes and translated; thus, transcription and translation events are coupled. The half life of PK mRNAs is very short, say 2-5 minutes. They are mostly polycistronic. Each of the mRNAs has a 5’ UTR region of 30 to 100 nucleotides before or at the upstream of Initiator codon called leader sequence. Just about 9 to 10 nucleotides left of the Initiator sequence or upstream of it, contains a sequence called Shine Dalgarno sequence “UAGGAGG”. The position and the sequence id very important. This sequence has complementary sequence at the 3’ end of the 16S rRNA of small ribosomal subunit. Interaction between them and base pairing leads to the binding of mRNA to small subunit of ribosomes during translational initiation. It has coding region starting from AUG and ends in a Terminator sequence. The size of the coding sequence varies from one gene to the other. Beyond or at the 3’ end of the nonsense codon there is another UTR sequence, perhaps without any function? The coding region is split into cistrons, where the mRNA generates more than one polypeptide chain. Between such coding sequences there are spacers. Thus, mRNAs are considered as polycistronic. That doesn’t mean there are no monocistronic mRNAs in bacteria.
5’---s/d-AUG------------------UGA---//--AUG-------------UAA------3’
Coding-1 spacer coding-2
Protein-1 protein-2
Shine Dalgarno sequence: AGGAGG
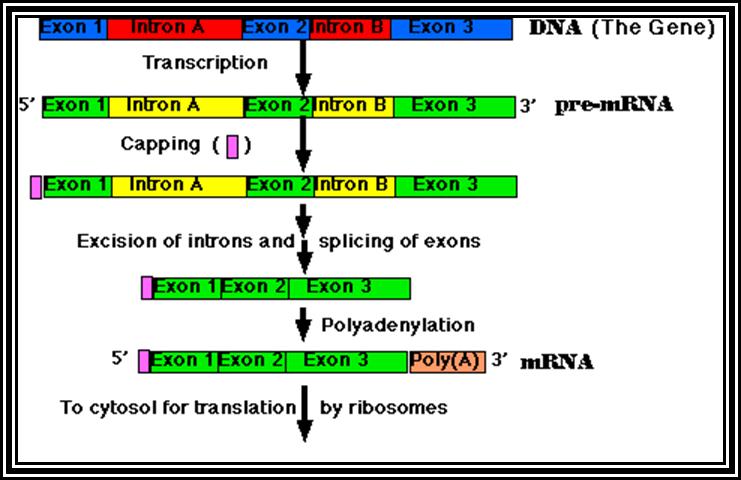
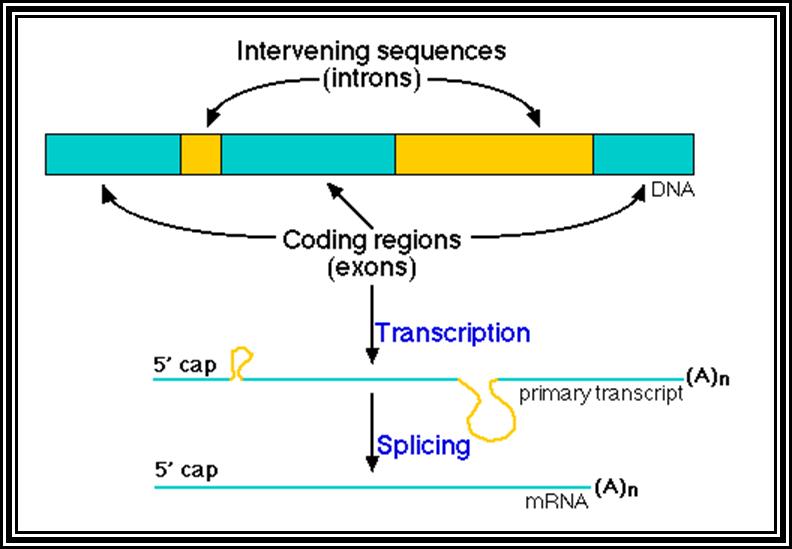
Eukaryotic mRNA: They are synthesized as pre-mRNA in the nucleus, then processed and transported into the cytoplasm, thus the site ant time of synthesis and translation are separated bin space and time. The pre-mRNAs are longer than the translating mRNAs. The pre-mRNAs or they are also called hn RNA (heterogenous RNA for their size and structures are different from one another), have coding region starts from an initiator codon and terminator codon. The coding sequence is divided into non-coding regions called Introns, mostly longer in size, and coding sequences called Exons. Thus the processed mRNA, in general monocistronic. The pre-mRNAs have a leader sequence (UTR) at the 5’ end and another UTR at the 3’ end. Most importantly the eukaryotic mRNAs have a sequence just upstream of the initiator sequence called KOZAK sequence 5’G/ACC.AUG.G3’ and an initiator sequence (InR) 5’pypy A/G pypy3’ at the site where the first nucleotide is incorporated during the synthesis of mRNAs. All most all pre-mRNA are subjected to processing, means they are modified, in the sense, the introns are cut and exons are spliced to generate full length translatable mRNAs. The 5’ end is added with 7’methylated GMP to the first nucleotide of the mRNA, it is A or G in a 5’ p-p-p 5’ A orientation. This process is capping; the caps can at different positions so they are called cap-O, cap-1, cap-2 and cap-3. Similarly, the 3’ end of the pre-mRNA is very long, but beyond the TER codons the mRNA has poly (A) signal sequence called AAUAAA. Using this sequence certain enzyme complexes cut the mRNA 28-35 nucleotides downstream of the signal sequence and adds 200 or more A's called poly (A) tail. Unprocessed mRNAs are not transported out of the nucleus. Nearly 70% of the pre-mRNAs are degraded within the nucleus. Thus the processed mRNA contains a cap at its 5’ end and a poly (A) tail at its 3’end. And the coding sequence is culled together exons.
7’CH3G5’-P-P-P-5A/G-NNN--------//----------AAuAAA----(A)n
Kozak sequence
5’cap---leader----A/G CCC AUG G------//------------UGA-----(A)n
5’cap--IAUG-exon-IiiiiiiiiiiiiiiiiI-exon--IiiiiiiiiiiiI-exon-UGAI-(A)n
Iiiiiiiiiiiii =Introns
I-------I =exons
Processing especially splicing, that is removal of introns and joining of exons in a predetermined is performed by a set of small molecular weight nuclear RNAs (snRNAs) and snRNA associated proteins (snRNPs). Introns and exons have splicing joints. The introns have specific sequences at 5’ splicing junction and 3’splicing point. They also contain branch point with a specific sequence and pyrimidines tract. These sequences are used by a complex of snRNA/snRNPs, and they bind to specific sequences and perform splicing, in the sense, they cut the 5’ splicing end first, where the cut 5’ end of the intron is covalently linked to the 2’OH of the A of the branch point. The 3’ OH of the first exon brings about the reaction where the 3’ end of the first exon joins with 5’ end of the next exon, thus the intron is released as lariat chain. Depending upon the way the introns are involved in splicng they are grouped into General (the above-mentioned process), Group-I intron splicing and Group-II intron splicing. Beside the general mode of splicing, tissue specific splicing takes place, where one type pre-mRNA is spliced in one way in one tissue and the same is spliced in another way in another tissue. But in some the mRNA produced has many sequences missing or extra, in such situations, nucleotides are added or removed, and the process is called editing done by specific editosomes. In some of the mRNA’s splicing is general but one of the nucleotides converted to the other in one tissue and in the other tissue splicing is general and with out editing.
Most of the mRNAs are monocistronic, but there are exceptions to this rule, where some animals do exhibit polycistronic RNA, eg. Caenorhabditis elegans and Trypanosomes, and some of them have common leader sequence. Some, though monocistronic the protein produced is polyproteins and the same undergoes proteolysis sequentially to generate small but functional polypeptides. Variety is the name of the game in EK.
Only those mRNA that are processed are transported into cytoplasm in association with ribosomes for translation, otherwise they remain in the Nucleus and there they get degraded.
Only 1.2 to 2 % of the total genome is involved in the production of mRNAs. The total number of mRNA species produced in a given tissue ranges from 15000 (intestine) to 22000 (brain). Among them nearly 12000 of them are common for all tissues, called house keeping gene products and rest of them are tissue specific, which actually determine the structure and the function of the said tissue. Each of the species of mRNAs have copy numbers ranging from 5 to 10 at any given time, but population of some can be 100 000 or more per cell, again that depends upon the cell type. However, some species of mRNA produced remain alive only for a short period of time and they are expressed only once in the life time of an organism.
Small molecular wt. RNAs: They are smaller in size found in the nucleus or in cytoplasm. Depending upon their location and function they classified into Sn RNA (small molecular weight nuclear RNA), Sc RNA (small molecular weight cytoplasmic RNA), sno RNAs, Tm RNA (tRNA like and mRNA like), Si or miRNA (small interference or microRNAs.
Sn RNA:
They are many such RNAs whose size ranges from 120 to 350 ntds. Some such RNAs are located in the nucleolus called sno RNAs. Each class of Sn RNAs is distinguished by their size and their function. Sno RNAs are involved in processing rRNA precursors. On the other hand, U1, U2, U4, U5, U6 and U7 Sn RNA are involved in pre-mRNA splicing. The U3 sn RNAs and U3-snRNPs are involved in splicing of histone premRNA species. Each of these sn RNAs are associated with common proteins and some species-specific proteins called sn-proteins (snurps). Some of these RNAs have ability to perform enzymatic functions.
Sc RNAs:
A good example for sc RNAs is 7sL RNA. Once it is synthesized, after processing it moves into the cytoplasm and gets associated with specific proteins called scRnPs. The sc RNAs have few domains bound by specific proteins. These are involved in transferring translating ribosomes on to endoplasmic reticular surface. This helps in transporting proteins into the lumen of Endoplasmic reticulum.
GENE CONCEPT
Classical view: Charles Darwin was the first biologist to visualize the presence of some particulates as heritable factors. Gametes were considered as the collection of such factors representing each and every part of the body. Such particulates of inheritance were called as Pan Genes.
Gregor John Mendel proposed laws of inheritance on the basis of unit characters or factors well founded by inheritance pattern observed in pea plants. Later Johansen called such factors as Genes, Thus, genes were called as units of heredity
The discovery of chromosomes as vehicles of heredity made Biologists to accept it as a fact that the genes are located in chromosomes, oblivious of the fact that chloroplasts and mitochondria contained hereditary material. Morgan Hunt demonstrated the presence of genes in chromosomes, where the genes are arranged linearly and linked to one another. Each chromosome is one linkage unit and human haploid genome consists of 23 such linkage units.
Modern view: Discovery of DNA as the genetic material has led to consider the gene as the unit of heredity capable of replication (replicon), crossing over (Recon), mutation (Muton) and function (Cistron). Beadle and Tatum’s' “one gene one function” or “one gene one enzyme” concept was changed by Benzer et al, as "one gene-one cistron or one cistron-one polypeptide".
Molecular definition: At molecular level, the gene is defined as a segment of DNA which is capable of producing a meaningful RNA or a meaningful polypeptide (s). Such a segment is called as unit of heredity, which fulfills the parameters such as unit of replication, unit of recombination, unit of mutation and and unit of function, which inherently is quintessence life molecule. Though it is true to greater extent, the present-day knowledge suggest that a single gene can produce more than one protein, more importantly individual genes need not be distinct entities, but they can overlap one another as overlapped genes. Recent work on nucleotide mapping of 6 X 174 viral DNA illustrates the fact that certain genes contain genes within the gene called overlapping genes. Further work on genetic DNA has revealed that the genes in eukaryotes are split and consists of noncoding (Introns) and coding (Exons) regions. In prokaryotes most of the genes generate more than one protein from a single transcript, thus it is considered as polycistronic. On the contrary eukaryotes have monocistronic features. The concept allele is nothing but the existence of alternate form of genes, which can bi allelic or multiallelic. A substantial number of genes are cryptic and remain hidden and express only under extraordinary stimulation. There are many genes which are called pseudo genes, which don’t function. Many such genes are processed genes, in the sense a processed mRNA is converted into ds-cDNA by reverse transcriptase and inserted into the genome. There is a class of genes, which live for themselves called selfish genes, for they code for their replication and transposition-called transposons, which include Retrotransposons. Though many genes are unique and exist as single copy per genome, there are genes exist as a family of genes, ex. Globin genes, Tubulin genes, Immunoglobulin genes. Some of the nonstructural genes like rRNA genes and 5sRNA genes and tRNA genes exist in multiple copies whose number ranges from 7 (in E. coli) to 200 to more than 100 copies in higher eukaryotes.
The total number of genes per a given genome can be approximately calculated based on the size of the protein it produces. In prokaryote the calculation on the basis of protein size and the size of noncoding regulatory region one can calculate the approximate size. But in eukaryotes, the size of the protein does not give a correct picture for the gene has an elaborate noncoding region such as introns, regulatory regions, spacers, and enhancer and activator regions. Taking only the size of mRNA determining the size is misleading. Yet approximate numbers can be derived from taking total length of nonrepetitive class of DNA and dividing this by average length of the standard mRNA. To illustrate let us take an example of haploid genome size is 1X10^9 base pairs. Nearly 25% of the genome is highly repetitive class which does not code for any known functional components, and another 30% is moderately repetitive class, which code for nonstructural components such as rRNA, tRNAs, sn/sc RNAs. The rest of the genome 45% amounts to 5.5 X10^8, it is this DNA that codes for proteins which constitute structural genes. Taking average length of an mRNA is 2000 ntds long, dividing 5.5X10^8 by 2X10^3 gives 2.75X10^5 genes. This number looks like an exaggerated number for the simple reason how can one account for the function of each of these genes. Present estimation of human genome i.e3X10^9, after deducting highly repetitive and moderately repetitive genes, it is estimated that there can be 1.5X10^6 genes, but the actual count based on the genomic library and sequencing all the expressed suggest that human may contain about 30000 to 33000 genes. This is big let down for those who expected at least a million gens in Homo sapiens. However the size of the gene varies from one gene to the other and from one species to the other. A single gene can as small as 350 bp to as big as 150000bp.
STRUCTURE OF THE GENE
Each gene, by virtue of being the unit of heredity, shows some structural and functional features. Irrespective of their size and source they do show some common features.
In general, every structural gene has a coding sequence, irrespective of structural components, which starts from an initiator codon and ends in a terminator codon.
Coding Block
5’-------5’AUG--------------------UGA 3’---------3’
On either side of the coding region, it has several noncoding segments and each of which with a specific sequence context and function. The size of these sequences varies from one gene to the other. The 5’end non-coding region contains sequences which act as promoters and 3’ end region is considered as terminal sequences. The promoter region is a regulator region at which transcriptional enzymes and the other required factors bind and initiate transcription at a site called START; this can be A or G and rarely pyrimidines. The terminal region beyond the TER codon contains sequences, whose size varies considerably, for the termination of transcription. There ends the common features for PK and EK.
* Leader
5’-------------A/G--------AUG----------//----UGA-----~~~~--3’
-promoter--- <……-coding…………> Terminal
* = Start point
Prokaryotic Gene Structure:
Promoters’ regions:

In E. coli and other prokaryotic systems, the promoter region has blocks of sequences upstream of the start. The transcriptional initiation point is considered as +1. On to the right side of the start nucleotides are referred to as down stream ntds and numbered as +2, +3 +++ etc. On to the left of the start the region is called upstream and numbered as -1, -2, -3, -20 etc. The length of the upstream sequence can range from 100 to 200 or more. The upstream region consists several sequence motifs; their sequence, size and location from the start varies from gene to gene but it is specific to a particular gene. The promoter of a gene is considered as a block of sequences which provide the structural motifs for the binding and assembly of Transcriptional apparatus and initiate transcription from a pre determined site called START point. The promoter region need not be upstream of the Start; it can be in downstream of the Start. Though promoter region provides the elements to the binding of RNA-pols, yet transcription may not be initiated, this can be due to a block next to the RNA-pol binding site which prevents the movement of the Enzyme; such substances are called repressors; or this can be due to the enzyme’s inability become active for transcription and requires another factor which binding to it another sequence contact the enzyme and make it active; they are called activators. There are elements which may be present at -200 or -100 or +1000 and when certain factors bind to these regions the rate of transcription and its efficiency increases by 100 to 200-fold, such sequences are called enhancers and the factors are enhancer proteins.
The most important and sine quo non sequence found at -10 is TATAAT box or Pribnow box. This is a consensus sequence found in almost all the promoters of all genes. This sequence is recognized by the components of RNA-polymerase and bind to it and initiate transcription at the start site. Without which RNA-Pol cannot firmly bind to DNA, open the DNA and initiate transcription.
I--- -200----I--I-- -65-I-- -35-I------- -10---I +1-
TTGACA TATAAT
The general consensus sequence in E. coli is -35 TTGACA and -10 is TATAAT. When RNA-Pol holozyme binds to TATAAT sequence it also covers the -35 sequence. Infect when the holozyme binds to this region it covers almost -65 to + 20 = 80-85 bp. However, few other genes have promoter sequences which differ from the general sequence blocks.
Coding region:
The coding region is not interrupted by non-coding segments. It starts from the AUG and ends in UGA or UAA or UAG. The coding region, though continuous, the transcript produced can generate more than one distinct protein, thus the transcript is polycistronic. Every cistron in the polycistronic mRNA has spacer region between cistrons. Each cistron begins with an AUG and ends in a TER codon, then a non coding spacer and again the next cistron begins with an AUG codon and ends in a TER codon; that is how polycistronic genes and their transcripts are built.
Gene terminal region:
The terminal region of the Gene has certain sequences beyond the TER codon, which contain segments of palindromic sequences of 60 -- 80 bp long, which on transcription produces an RNA terminal region consisting of 57 nucleotides with some GC rich sequences and terminated with an A and five U s . As the enzyme reaches this region it generates a stem loop structure followed by an A and followed by 5 Us. The stem loop structure somehow stalls the enzyme, and the Us pair with A are weekly base paired the RNA dissociates from the template, thus the transcription is terminated. Nevertheless, the sequences rich in Cs, and they are involved in the termination transcription by what is known as Rho dependent termination.
Eukaryotic Gene Structure:
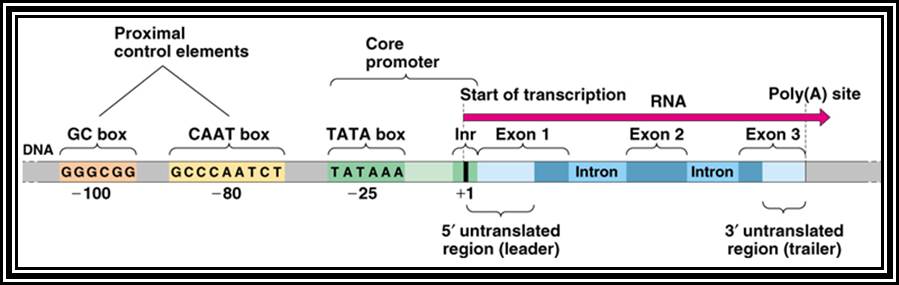
Eukaryotic gene structural organization is built on the same pattern as that of prokaryote, structurally it shows lot of variation among the group of genes for many genes are expressed in tissue specific manner, but majority of the genes expressed are common to all cell types, such genes are called house keeping genes. They express in all tissue types and at all times. Tissue specific expression, stage specific expression, and expression in response to stimuli requires different regulatory elements for either gene expression or gene repression or call it inactivation.
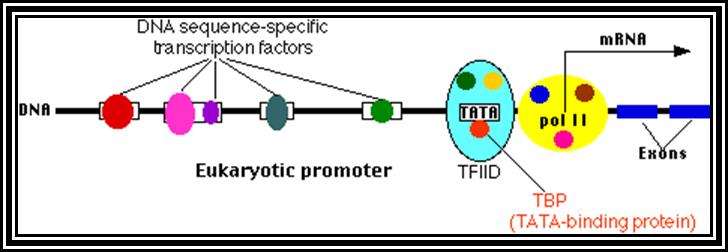
All genes contain a coding region, which is split; on the upstream of the of the coding region it has promoter region. Beyond the coding region of the gene, it has sequences for the termination of transcription and also in some cases it contains regulatory sequences for the stability. The upstream region is conventionally called promoter. By definition promoter is the site for the assembly of transcriptional apparatus and its accessory factors, where they bind to it and open the DNA to initiate transcription. The components of the promoter, depending upon the type of the gene, vary and the location of the promoters also varies. In general, -25 to -30 from the start, a consensus sequence is found called TATA or Hogness box, which acts as first recognition sequence for the assembly of RNA-pol complex, without which enzymes won’t assemble. The start point itself is bracketed by a set of sequences; hence this region is called InR box or InR sequence. The sequence of InR is -3py -2py -1C. +1A +2N +3T/A +4py +5py. In the upstream region of the TATA box, at a distance there several sequence boxes such as GC box GGGCGG at -90, a CAAT box at -75 and an eight base Octamer box. The number of such sequence and the position from the start or TATA box varies. Besides the binding of RNA polymerase complex to TATA box, other factors binding to specific sequence boxes increase the efficiency of transcription.
The promoter components vary depending upon the type of the RNA-pol that is involved. Eukaryotes have three different class of RNA-pols, such as RNA pol-I, pol-II and pol-III and each of them transcribe specific groups of RNAs, like rRNA genes by RNAP-I, structural genes by RNAP-II and tRNA, 5sRNA genes and few sn/ScRNAs by RNAP-III. In many cases the promoter elements are found located with in the gene itself, for example 5S RNA gene and tRNA gene.
Gene Structural Features:
Promoter coding Terminal
I<------->InR+>---I<AUG--------------//--------------UGA>I--------

Promoter Elements in General:
+1 START
I---OCTA-I---GC-I----CAAT---I------TATA------InR------>
Though the above description of the promoter is a general one, where both TATA and InR present; but there are many exception to this rule. A large number of genes including house keeping genes TATA less and InR plus promoters, or TATA less and InR less promoters.
Coding Region:
The coding region as in the case of prokaryotes starts with an initiator codon and ends in a nonsense codon where translation is terminated. The coding region in eukaryote is not continuous as in prokaryote, but the coding region in the newly synthesized mRNA, is interrupted with noncoding intervening sequences called introns. The coding segments are called EXONS and noncoding regions are called INTRONS. In most the genes the size of the introns far exceeds the size of exon. The coding exon in fact represents a motif of a protein. If one looks at the features of exons and structural motifs of the proteins, one can observe that almost all cellular proteins are made up of some few exons in combinatorial mode. It means, though there are more than 50-60,000 proteins, inspite of the limited number of existing exons, by mix and match combinations of exons, they have generated such huge number of proteins. The newly synthesized pre-mRNA is subjected a meticulous and programmed cutting and joining process where the introns are removed and exons are joined in a sequence; the process is called splicing. The size of the coding region varies depending upon eh size of the protein. However, most of the mRNAs are monocistronic, with few exceptions. Many a times the single protein produced is cleaved in tissue specific manner or stage specific manner and can generate functional proteins.
Gene terminal region:
The region of the gene beyond the TER codons is the terminal region whose size varies considerably. The structural features of terminal regions differ from rRNA genes, structural genes and tRNA/5sRNA genes and sc/sn RNA genes. Though the terminal region of rRNA genes resemble to that of prokaryote, but not the same, so also with that of the gene transcribed by RNAPs-III. However, the terminal region of structural genes is longer and elaborate. They have poly-A signal sequences and sequences like GU/C sequences for the recognition by specific proteins which cut the transcript at particular site and add poly (A) tail. In many mRNAs the 3’ noncoding region has certain sequences which generate certain structural features, which determine the stability of the mRNAs.
GENE EXPRESSION
According to the classical concept, the gene is always referred to a phenotype; like Red gene to Red color, Tall gene to Tall character. But the present-day knowledge about Gene clearly suggests that the primary function of a gene is to produce either RNAs or polypeptides through mRNAs. As polypeptides are functional molecules, they bring about specific reaction; and the products, thus express the characters; be it a biochemical or a morphological one.
As there are different classes and kinds of genes, some genes produce rRNAs, some produce tRNAs, sn RNAs, etc and others produce mRNAs. Only the mRNAs carry information for the synthesis of a polypeptide. Though other RNAs do not carry any message for proteins they are involved in protein synthesis in one way or the other. In this context it is important to remember that the gene expression involves two distinct but related processes such as Transcription (RNA synthesis) as the first step and Translation (Protein synthesis) as the second step. Both these stages are regulated.
Initiation of RNA synthesis is referred to as Gene activation (Switch on) and the repression or inhibition of RNA synthesis is called gene inactivation (switch off).
Translation and production functional protein as the end product is also the part of gene expression. It is ultimately the protein or the combination of protein reactions results in a phenotypic expression. Thus, one cannot simply assume one gene produces one phenotypic character. A single character like color of the flower is determined by at least six or seven gene products, though the last reaction produces phenotypic character. The concept of single gene attributing to single phenotype is a misconception and wrong. It is perfectly OK if one considers the protein (gene product) involved in the last step of the multistep reaction resulting in a phenotypic expression, yet it is one step in whole multistep reactions.
Gene Expression in Prokaryotes- First Step is Transcription:
The synthesis of RNAs requires specific DNA template, all ribonucleotide triphosphates (rNTPs), inorganic metal ions like Mg^2+ or Mn^2+ and other factors. In contrast to eukaryotes prokaryotes have only one kind of RNA polymerase. The DNA template of a structural gene to be transcribed consists of a promoter consisting of TATAATA region at -10 and TTGACA at -35, these are the basic requirements for gene to be expressed. The process requires enzymes, and few other factors. Transcription takes place in three stages in sequence named as Chain Initiation, Chain Elongation and Chain Termination.
Promoter region: I-- -100--- - 65---- -35--- -10------+1>---------
During transcription only one of the two strands is used for, the strand used is referred to as (-) strand, the other strand is called (+) strand for the sequence of the (+) strand is same as that of mRNA. Two genes which exist side by side, In one of the gene the lower strand acts as the template for transcription, for it promoter is on the left part of the gene. The gene at the side of the first has promoter on the right side of the gene but both genes are components of the same DNA. In the gene -2 the strand used as template for transcription can be the top strand. For any given segment of the DNA which acts as the Gene only one strand is used as the template and not both.
Gene-1 3’I<~~~~~~~~~~~~~I5’
(+) 5’I-P-I-------------------I-I------------------------I--P-I (-)
(-) 3’ I-P-I------------------I-I------------------------I-P--I (+)
5’ I~~~~~~~~~~~~~~>I3’ gene-2
+/- = double stranded DNA.
P = promoter.
5’~~~> = Transcript- direction
I-I = spacer between two genes.
1. Chain Initiation :
Chain initiation requiers RNAPol holozyme complexa. The core enzyme consists of multiple subunits like two alpha subunits 36-40KD), Beta subunit (150-155 KD), one b’ beta prime subunit (155-160 KD). It also consists of omega 11KD, which is not actually required. The most important component is the sigma factor (70KD); all together acts as the Holozyme. The Beta subunit has catalytic site for polymerization, beta prime has DNA binding site. They also contain metal ions Zn2^+ binding site. Together they have polymerization property in %’> 3’ direction. Two alpha factors are little longish and facilitate in the assembly of core enzyme and also helps in recognizing the -35 region of the promoter. The sigma-70 is the most common factor whose association determines which gene to be transcribed and which need not be transcribed. Most of the house keeping genes and many more genes are recognized by the sig-70 and transcription is initiated in them.
|
Protein |
Gene |
Mol.wt(KD) |
Subunits |
Function |
|
Alpha |
Rpo- |
36-40 |
2 |
Assembly of the core complex |
|
Beta |
Rpo-B |
150-155 |
1 |
Catalytic |
|
Bete’ |
Rpo-C |
155-160 |
1 |
Binds to DNA |
|
Omega |
Rpo-Z |
11 |
1 |
Regulates the growth of the cell |
|
Sigma |
Sig |
70 (can vary) |
1 |
Identifies the promoter and activates the the pol |
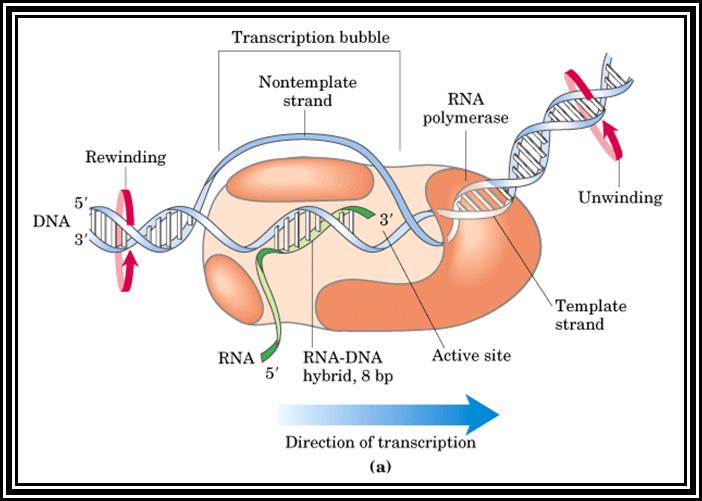
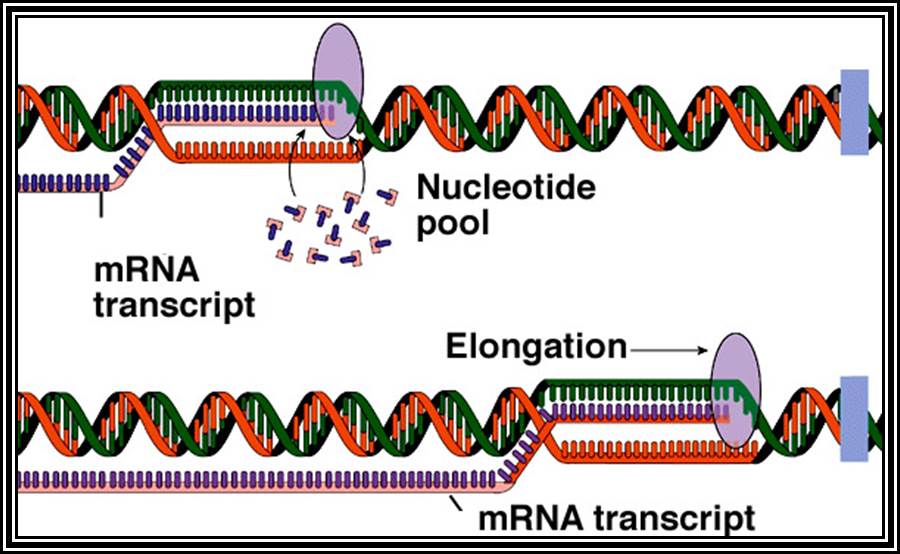
Sigma factor is a longish protein with many domains. In association with RNAP-core complex, when it climbs on to DNA, the complex diffuses till it comes in contact with TATAATA region and halts. At this site if it has -35 TTGAC it binds. This sequence is the recognition site for sigma-70. There are at least 17 addition sig factors which recognize different -35 sequences and initiate transcription. Once the TATAATa and TTGAC are regnized by the sig-70, it activates the core enzyme. Now th enzyme binds tightly to the promoter element and unwinds the DNA 15-18 base pair long into ss DNA templates as transcriptional bubble During this process the Holozyme goes through some isomerization steps such as tight binding and loose binding and again tight binding. Then the enzyme start initiating the incorporation of the nucleotides at predetermined site called Start site. The TATAAT site provides identity and specificity for the initiation sites. If the sequence of TATAATA site is changed, though polymerase initiates trascription but at different site away from the TATAAT box. Once the initiation complex is formed with first nucleotide at the first site, another ribonucleotide settles by the side of the first nucleotide at the elongation site. Then the polymerase brings about the bond formation between the 3' OH group of the first nucleotide and the 5' OH group of the phosphate group of the second nucleotide. Initiatially assembly of nucleotides goes unto 5-7 and fails to continue. Again it reinitiates, like this it make few attempts, and when it produces a length of 10-12 ntds long RNA then it starts elongating.
2. Chain Elongation: polymerization of nucleotides is overcomes, the enzyme now moves ahead of the promoter region, this event are called promoter clearance, which actually the elongation process. The breakdown of the phosphate at alpha position and breakdown of the pyrophosphate bonds provides the energy for the phosphodiester bond formation as well as the energy for RNA polymerase movement over the DNA strand and the polymerization of RNA nucleotides progress. At the same time the enzyme moves into the fork ahead and continuously unwinds the DNA ahead of the fork. As the movement of the enzyme complex move forward with polymerization of complementary nucleotides, the opened DNA in the back side of the fork rewinds into helix, thus the newly synthesized RNA is threaded through the enzymes exit tunnel. To start with the enzyme occupies from -60 to +20 region, but as it progresses with the synthesis of RNA it contracts and moves like an inchworm mode. Once the enzyme clears the promoter region the sigma factor dissociates and few other factors like Nus and others associate, which are required for elongation. The Nus and sigma association is mutually exclusive.
2. Chain Termination: When RNA polymerase reaches the end of the gene, at the termination site it generates a nontranscribable region. The termination takes place in two modes; one is through the generation of stem loop structure followed by a four Us. In another the transcription is terminated by motor protein called Rho. In majority of the case transcription is terminated by stem-loop mode. The last part of the transcript generates a stem loop because few complementary sequences which are GC rich, then the loop follows few Us. As the hydrogen bond between As and Us is so weak the transcript falls off from the template. First the stem loop structure halts the progress of the RNAP and then the transcript falls off. This leads to the dissociation of the RNAP from the template to reassociate elsewhere to initiate another transcription in another gene. In case of Rho dependent termination, the Rho is an hexamer and it associates with the 5’ end of the transcript and move in AT dependent manner and when the RNA-pol reaches the end of the gene it generates a sequence, which is recognized by the Rho factor and halts the progress of the RNAP and dissociates the transcript from the template and the enzyme.
Once the region of the promoter is open for initiation of transcription is repeatedly used for initiation after initiation as a cascade of initiation and process initiation and elongation and termination become continuous cycles.
The process of RNA synthesis is highly regulated by many intrinsic and extrinsic factors. The rate of RNA synthesis varies from 1000 nucleotides tor 2000 per minute. It also performs proof reading during transcription.
In prokaryotes transcription and translation events are coupled for the half life of mRNA is hardly 2 to 5 minutes. Immediate initiation of translation prevents immediate degradation by exonucleases in the cell.
Transcription in Eukaryotes:
Eukaryotic promoters have varied structural modules. Though basically the upstream of start nucleotide contains TATA box at -25 to -35, there few more modules such as CCAAT box at -70 and GGGCGG box at -90 and further up there can be structural motifs such as Octamer and further more there can be enhancer element also. The number and location of each of theses modules vary from promoter to promoter. In some cases the promoter are located within the coding region itself.
Whatever might be the modules and positions they are recognized by Transcriptional apparatus, which binds promoter elements recognizing the TATA as the identifying site. It is the site that ultimately determines the transcriptional initiation point.
The transcriptional apparatus consists of core complexes and accessory complexes. The components these complexes, depending upon the kind of RNA polymerase, vary from one another. The RNA pol-I transcribes ribosomal RNA genes. RNA pol-II transcribes structural RNA genes and few sn RNA genes. Te RNA pol-III transcribes 5sRNA genes, few sn/sc RNA genes, sno-RNA genes and tRNA genes. The promoter elements of each of these genes have their own specific promoter elements and structural components of each of these transcriptional components differ from one another.